Az ARG1-D tünetei más neurológiai és neurometabolikus betegségek tüneteihez hasonlóak, mint például egyéb karbamidciklus zavarok (UCD), cerebral paresis (CP) vagy herediter spasztikus paraplegia (HSP)5,6.
Az ARG1-D elkülönítő diagnosztikájához a magas plazma argininszinttel4-7 járó klinikai tünetek azonosítása szükséges.
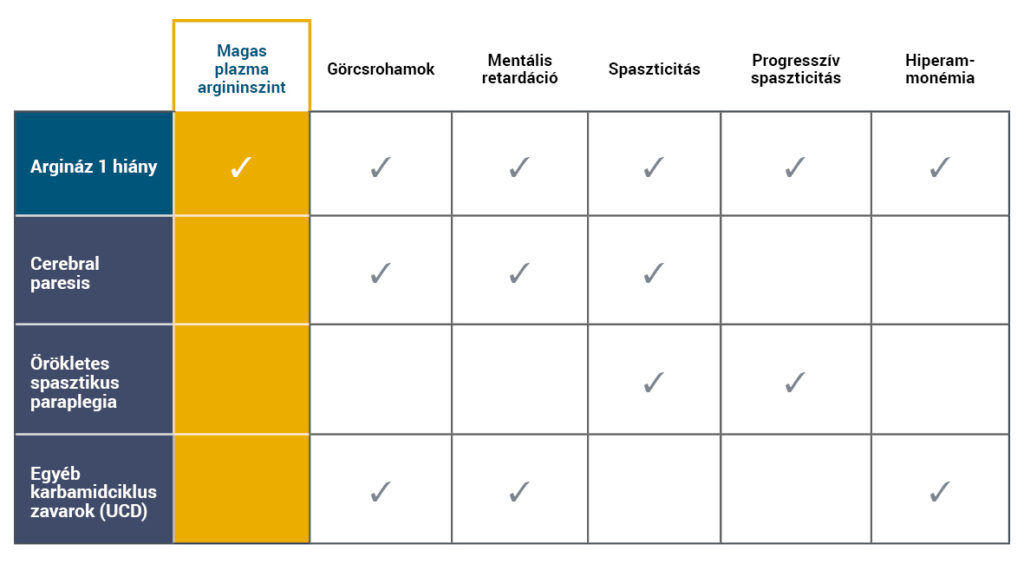
- A hiperammonémia nem jellegzetes tünete az ARG1-D-nek, ebben a kórképben akut hiperammonémiás epizód ritkán fordul elő4,8.
Az újszülöttkori szűrés korlátai miatt az ARG1-D-t gyakran nem diagnosztizálják:
- A szűréshez szükséges arginin határérték meghatározása nem egyértelmű az arginin maternofoetális transzportja miatt9,10.
- A szűrési algoritmusok és az arginin határértékek eltérőek9.
- Az ARG1-D nem szerepel az európai országok többségének újszülöttkori szűrésében11.
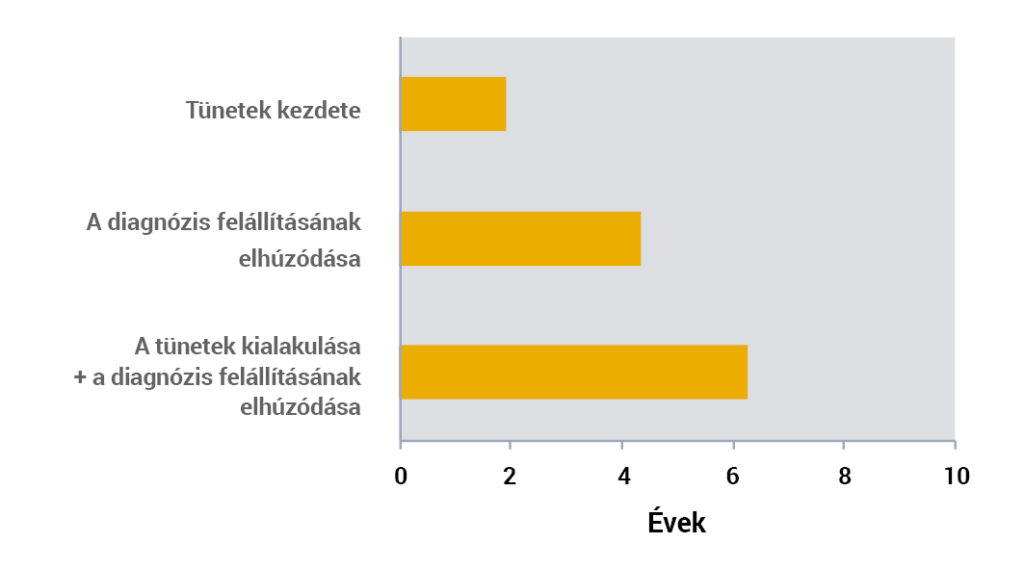
A késői diagnózis és a tünetek késői jelentkezése miatt a kezelés megkezdése általában ~6 éves korra esik1.
A plazma aminosav profil meghatározás és a genetikai teszt elvégzése megerősítheti az ARG1-D diagnózisát12,13.
A diagnózis felállítása előtt fontos a teljes egészségügyi-, táplálkozási-, családi- és szociális előzmények felvétele és az alapos fizikális vizsgálat elvégzése.
Az ARG1-D tüneteiért felelős magas argininszint célzott anyagcsere vizsgálattal kimutatható3,12,13.


Magas plazma argininszint esetén genetikai vizsgálat† szükséges a diagnózis megerősítéséhez.
†Az ARG1-D genotípusának heterogenitása miatt nem azonosították az összes betegségokozó mutációt.
Hivatkozás:
1. Huemer M, et al. J Inherit Metab Dis. 2016;39:331-340. 2. Edwards RL, et al. J Inherit Metab Dis. 2009;32:S197-S200. 3. De Deyn PP, et al. Hyperargininemia: a treatable inborn error of metabolism. In: Guanidino Compounds in Biology and Medicine. London, UK: John Libbey Company Ltd; 1997:53-69. 4. Burrage LC, et al. Hum Mol Genet. 2015;24:6417-6427. 5. Carvalho DR, et al. Pediatr Neurol. 2012;46:369-374. 6. Prasad A, et al. J Child Neurol. 1997;12:301-309. 7. Crombez EA, Cederbaum SD. Mol Genet Metab. 2005;84:243-251. 8. Scaglia F, Lee B. Am J Med Genet C Semin Med Genet. 2006;142C:113-120. 9. Therrell BL, et al. Mol Genet Metab. 2017;121:308–313. 10. Pitt JJ. Clin Biochem Rev. 2010;31:57-68. 11. Loeber JG, Platis D, Zetterström RH et al. Int J Neonatal Screening. 2021;7:15. 12. Sun A, et al. Arginase deficiency. In: Adam MP, et al, eds. GeneReviews®. Seattle, WA: University of Washington, Seattle; 2020. 13. Ah Mew N, et al. Urea Cycle Disorders Overview. 2003. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1217/. Accessed November 26, 2021. 14. Cai X, et al. Medicine (Baltimore). 2018;97:e9880. 15. Bélanger SA, et al. Paediatr Child Health. 2018;23:403-410. 16. Lüneburg N, et al. J Nutr. 2011;141:2186-2190.